Scale-up reactions account for a number of accidents every year due to the increased hazards compared with smaller scale reactions. The purpose of the following sections is to help identify and control the hazards associated with scale-up reactions and to create a more safe and efficient synthetic route.
Reaction Risk Assessment
If the reaction is being conducted for the first time, or the scale-up reaction has potentially dangerous bonds present, fill out the Risk Assessment for Chemical Experiments worksheet before starting the experiment. Follow the steps below until you are confident the reaction can be performed safely.
- Determine if the hazards identified in the Risk Assessment and Potentially Explosive Experiments are more dangerous on large scale (e.g., hydrogen gas as a by-product).
- Review the scientific literature, web sources, SDS’s, Brethericks Handbook of Reactive Chemical Hazards, and consult resident experts to identify hazards.
- Are these reactions known to be dangerous and what controls can be utilized to mitigate the dangers?
Scale-up Reactions
Every reaction must be assessed before scale-up to determine if there is any potential for uncontrolled events. Start hazardous reactions small and increase the scale by a maximum of three-fold for each scale-up. Diligently watch for warning signs and reaction rates each time. More mundane reactions can be scaled up by more than three-fold (e.g., EDC couplings, base hydrolysis, Fischer esterification). If any reaction conditions or reagents change, restart the reaction over again at a small scale to test its safety. Avoid changing reaction conditions and reagents in the middle of an experiment.
Scale
- Small: Reactant and solvent amounts: <1 gram of substrate, solvent < 25 mL.
- Moderate: Reactant and solvent amounts: 1-15 grams of substrate, solvent 25 – 500 mL.
- Large: Reactant and solvent amounts: >15 grams of substrate, solvent > 500 mL.
Communication
Good communication is essential when performing reaction scale-ups. Talk with others who have done the experiment before – they may have forgotten to record some critical detail(s) about the experiment. Inform anyone working nearby who might be affected if the reaction gets out of control.
- Notebooks must be sufficiently clear and legible (with detailed information) to allow someone else to perform the experiment.
- Include details on the solvents and reagents used, such as purity and manufacturer.
- Record any concerning adverse events, observations, dangers, and other important facts such as induction periods and runaway reactions.
- Provide an explanation for why a certain technique is critical. An example is the use of a magnetic stir bar as a heat sink during drop-wise additions and how it affects the formation of side products. Always alert others in the area when your reaction emits substances that could react with other substances; e.g., emission of hydrogen gas when others are working with pyrophorics; emission of NH3 gas as others are emitting HCl in chemical fume hoods that could lead to NH4Cl build-up in duct work.
- Alert others when you are working with or creating compounds that contain hazardous bonds (e.g., large scale nitration reactions, cyanohydrin reactions, diazomethane generation). These reaction scale-ups can affect others in the area if they are not contained by the fume hood.
Time
The bigger the reaction, the longer the setup, run, and workup of the reaction will take. Allot extra time for the experiment, and do not try to rush the reaction.
Temperature
Temperature control is critical for safely performing scale-up reactions. Remember basic reaction kinetics. As the temperature increases, so will the reaction rate and therefore produce more heat. This additional heat then feeds into a positive feedback loop and can cause a reaction to spiral out of control. Loss of temperature control can have devastating results and cause side-products to form.
- Use a thermocouple probe or thermometer to monitor the internal reaction temperature, which can differ significantly from the temperature of the oil bath outside of reaction vessel.
- If heat generated from reaction is not controllable using standard methods, do not scale up further.
- Determine if the cooling method you have chosen is sufficient for the scale you will be working with. Have a backup plan in case it fails.
Stirring
Use overhead stirrers for consistent stirring of larger reactions to prevent hot spots. Magnetic stir bars do not mix large, thick mixtures well, and overhead stirring is more consistent when scaling up from one level to the next.
Monitoring Reaction
Monitor the reaction frequently for warning signs. Be alert to equipment failures (e.g., condensers not working, leaks in glassware joints, obstructed stirring), gas evolution, color change, viscosity differences, and temperature spikes.
Equipment
Choosing the right equipment for reaction scale-up can make a major impact on the efficiency and safety of the reaction to be performed.
- Use Teflon sleeves to prevent leaks and facilitate glassware disassembly.
- Use wide mouth glassware to facilitate entry and removal of materials along with good venting if emergency arises (massive gas evolution).
- Use lab jacks to elevate vessel heat sources so that they can be removed quickly if thermal runaway occurs.
- The volume of the vessel should be at least twice the volume of all added substances (including quenching material). Leave enough headspace in the event the reaction gets out of control.
- Avoid oil baths for large scale reactions – use heating mantles with probes. Oil baths have limited capacity and can create oil spills from equipment movement (sloshing).
- Avoid needle use for gas in-letting/out-letting on scale up reactions. This leads to excessive pressure increases. Rather, utilize gas inlet (or vacuum) adapters for gas lines. Adapters allow for better gas flow and venting if gas evolution increases rapidly.
- Make sure the fume hood ventilation system is sufficient for the scale you are working with. Larger equipment takes up more volume in the hood and can affect air flow. Be sure large equipment will not interfere with the chemical fume hood’s ability to operate properly.
Solvent and Reagent Selection
Solvent and reagent selection can have a critical effect upon reaction scale-up safety, especially if things go wrong. Here are a few general things to consider:
- Pay attention to the age of reagents and solvents–is the concentration of that old HCl bottle really 12M?
- Do not use materials from unlabeled containers.
- Be careful using materials from different lots (e.g., 5 grams from lot #A, 2 grams from lot #B, and 5 grams from lot #C).
- Be careful using materials from different manufacturers (e.g., previous reactions were done using vendor A materials but you intend to use vendor Z this time). Purity levels vary amongst manufacturers.
- If changing reagents or solvents is unavoidable, perform a front-run first. That means first performing the reaction on a small scale and comparing the results with past reactions.
Solvents
Be aware of the flammability of solvents around the equipment you choose, and take the appropriate precautions.
- If possible, choose solvents that will boil before the product decomposes. This helps to prevent the formation of tar, impurities, and other side products.
- Be aware of the dangers (explosions) from concentrating peroxide-forming solvents. When working with >1000 ml of peroxide-forming solvent, check peroxide levels when the volume of solvent has been reduced to 10-20% of the original volume.
- Use freshly distilled or purchased solvents, especially if anhydrous conditions are needed.
- Avoid highly volatile solvents with low boiling points and peroxide-forming issues. Use of these solvents poses fire and explosion hazards, especially during operations involving transfers, heating, and concentrating. Highly volatile solvents are also prone to concentration changes (e.g., running the reaction vessel dry).
- Avoid THF and use 2-MeTHF (less peroxide issues, slightly higher b.p., and more environmentally friendly);
- Avoid Et2O and use t-BuOMe (less volatile, higher b.p., less peroxide issues);
- Avoid hexanes and use heptanes (higher b.p., less toxic, less volatile)
- Avoid CH2Cl2 & CHCl3 and use 1,2-dichloroethane (higher b.p., ALL halogenated solvents have toxicity issues).
- When performing air-sensitive reactions (e.g., Suzuki, Buchwald) sparge the solvents (run a steady stream of bubbling inert gas through the solvent via a 12-18” needle while in the reaction vessel). This is safer and works better than degassing with vacuum (freeze-pump-thaw method). Sparge solvent mixture for 30 minutes with stirring before adding sensitive reagents, then sparge for another 5-10 minutes after addition but before heating.
Reagents
Reagent quality can vary greatly from one manufacturer to another.
- Impurity profiles vary from lot to lot and from manufacturer to manufacturer. Perform front-runs first to verify results before proceeding.
- If using large equivalent excesses of a reagent, try running a more concentrated reaction with less equivalents.
- Use reagents that allow for better stoichiometric control and safer chemical handling and storage. Examples:
- Use NH4Cl and base rather than NH3 dissolved in Dioxane.
- Use NH4NO3 and H2SO4 rather than HNO3 and H2SO4
- Use dimethylamine hydrochloride and base rather than dimethylamine dissolved in water, THF, or methanol.
- Use TMS diazomethane or generate diazomethane in diethyl ether rather than distilling diazomethane.
- Use Oxalyl chloride/DMF (cat.) to create acid chlorides rather than using thionyl chloride.
- Avoid sensitive intermediates. Carefully determine how you will drive a reaction to completion. For example, with amide formation, it is easier to add more EDC, HOBT, and carboxylic acid than make more acid chloride if the reaction does not go to completion. Use reactions that will allow you to easily add more reagents (to drive reactions forward) rather than having to create more of an intermediate.
- If possible, use reagents that can make workups and purifications cleaner and easier. This can lead to cleaner reactions, less intensive purifications, lower flammable solvent usage, lower fire hazard, and less expenditure of time. Ultimately, efficiency and safety are increased. Examples:
- For Mitsunobu or Wittig reactions, use Me3P (1M solution) rather than Ph3P; Me3P=O is water-soluble and can be washed away; Ph3P=O is sometimes difficult to remove during purification.
- Use N,N-Dimethylethylenediamine (CAS# 108-00-9) to scavenge excess acid chlorides, acrylates, mixed anhydrides. Then wash away with a simple acid wash during workup.
Figure 1. N,N-Dimethylethylenediamine
*A Note Concerning Air- and Water-Sensitive Materials
- When using air- and water-sensitive materials, always be sure to properly inert your reaction vessel. It also helps to reduce the chance of fires with flammable solvents. Insufficient inerting is a recipe for disaster!
- Consider using a less hazardous reagent (substitution) or in a less hazardous form (attenuation). Examples:
- Hydrogen transfer agent rather than Raney nickel;
- Use n-BuLi rather than t-BuLi, if possible;
- Use NaH 60% dispersion in mineral oil rather than NaH, dry, 95%;
- Use 10% Pd on carbon (wet) rather than 10% Pd on carbon (dry);
- To prevent explosions and fires from chemical dusts (especially air and water sensitive dusts), use them in the form of slurries.
Runaway Reactions (Thermal Runaway)
Definition (Wikipedia): “A situation where an increase in temperature changes the conditions in a way that causes a further increase in temperature, often leading to a destructive result.”
- Determine if a reaction can generate significant heat.
- If possible, replace highly reactive reagents with less reactive ones.
- Use proper volume of solvent for sufficient heat transfer.
- Do not use large equivalent excesses of reagents.
- Watch reaction concentrations (on average should be between 1M–0.1M).
- Use heat sinks if possible.
- Avoid “neat” reactions on large scale.
- Ideally choose a temperature that allows the reaction to proceed efficiently (good rate with minimal side product formation) and is easily controllable below the solvent’s boiling point or is easily cooled (e.g., 0oC–55oC).
- Assure constant reactant supply by choosing solvents that will not freeze (e.g., HOAc @ 0oC) at reaction temperatures, and be sure all reactants are soluble in the solvent. Be careful of heterogeneous mixtures that may dissolve unevenly in solvent.
- Monitor internal reaction temperature (via probes) during additions. Ensure constant mixing to prevent reaction hot spots.
- Ensure that the addition rate does not exceed the reaction vessel’s ability to dissipate heat or relieve pressure.
- Never add additional reagents if the optimal reaction temperature is not being maintained.
- If refluxing, do not rely on a condenser as the only means to remove heat from the reaction. Use Lab Jacks to remove the heat source quickly from the reactor vessel.
- Cooling is mandatory if heat generated by the reaction exceeds the vessel’s heat transfer ability.
- Always have a means to cool the reaction vessel if the reaction gets out of control, e.g., pull the heat source and cool in a water bath.
Good temperature control (albeit addition rates or cooling) is key to preventing thermal runaways!
Induction Periods
Definition (IUPAC compendium of Chemical Terminology): “The initial slow phase of a chemical reaction which later accelerates.”
Many reactions that have induction periods quickly turn into runaway reactions.
- Is there evidence of an induction period from previous reactions? Nitrations, free radical reactions, and Grignard formations all have been known to have induction periods.
- If there is evidence of an induction period:
- Develop a strategy to control it (e.g., addition rates, cooling);
- Have a cooling method ready before initiating the reaction;
- Monitor the reaction closely, watching for signs of pressure, heat, color, gas evolution, solvent viscosity, and do not get complacent, regardless of the time involved.
To control induction periods via addition rates, add 5-10% of the reactant and watch for reaction initiation. If initiation is observed, carefully add the rest in a controlled manner. If it does not initiate, add another 5-10% of reactant and monitor. Remember the more reactant that is added, the greater the chance a runaway reaction will occur once the reaction initiates! Be patient and have a cooling system ready.
Workups
Scale-up reaction workups can pose many safety issues not normally encountered during smaller scale work. A few issues to consider:
- Be sure you have the right sized equipment for the scale, especially the use of separatory funnels and the number of extractions needed. Remember basic extraction rules–it is better to use more extractions with less solvent than fewer extractions with larger solvent amounts.
- Large equipment also poses physical constraints due to larger amounts of solvents, washes, and materials. Multiple extractions of dichloromethane on a large scale can not only pose physical difficulties on personnel but on equipment, e.g., ring stands, clamps, monkey bars.
- Cleaning large glassware after workup in sinks designed for smaller glassware can easily lead to broken glassware, which may cause lacerations.
- Workups on scale take longer to accomplish due to the scales involved. Quenches, extractions, and rotovaping of highly flammable materials on scale all pose major hazards that must be approached carefully and methodically.
- Quenching reactions (especially with reactive reagents) on scale must always be approached with special care. Allow the reaction to fully cool before quenching. Take your time and be thorough.
- Take the time to analyze each layer to be certain where your desired product is. Don’t dispose of any waste until you are satisfied with your yield and purity.
- When using peroxide-forming solvents on scale, be certain to check peroxide levels when solvent levels are reduced to ~10-20% of the starting volume.
Purification
If possible, avoid chromatography when doing scale-up synthesis due to the fire hazards of large amounts of flammable solvents. The size of specialized glassware (e.g., columns) can make for difficult handling and still may not give efficient separation of eluting bands, making it necessary to repeat chromatography procedures. Try crystallizations, extractions, triturations, and washes to minimize solvent usage.
Washes
Use standard and multiple washes of aqueous solutions (e.g., 5% NaHCO3, 1M citric acid, 5% brine, 1M NaOH) and analyze (via HPLC) both the wash and the organic layer to see how much and where impurities and desired products are going. Do not make assumptions about where they are; you might be shocked by the results! TIP – this is what good process chemists do!
Precipitations
Running reactions in water-miscible solvents (e.g., acetone, ethanol) makes precipitations ideal by the simple and slow addition of water. Be certain to analyze the solution to determine the amount of residual product still in solution. Tip - to maximize precipitation of product and increase the yield significantly, reduce the temperature of the mixture to 1-2oC before filtration.
Crystallization
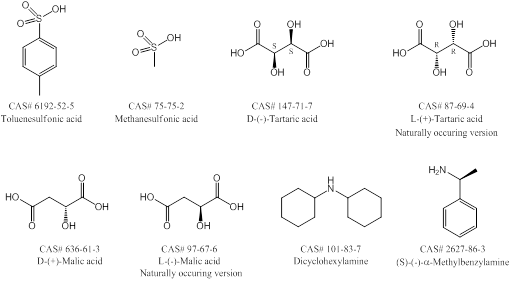
Use standard crystallization methods with single or multiple solvents. Forming salts of the desired product can help with crystallizing products. Screen multiple solvents and solvent combinations (using HPLC if available) to determine which solvents are best for solubilizing the product – this can also tell you which solvents solubilize the product the least and are therefore a good co-solvent. A few examples for salting are listed below in Figure 2.
Basic product: Use HCl, HBr, TFA, HOAc, methanesulfonic acid, toluenesulfonic acid, tartaric acid (especially for chiral crystallizations), etc.
Acidic product: Use NaOH, KOH, dicyclohexylamine, triethylamine or (S)-(-)-a-methylbenzylamine (especially for chiral crystalizations). TIP – dicyclohexylamine is a favorite because it almost never fails.
Absorbent materials
Activated carbon is a highly efficient material for removing impurities, and it comes in a multitude of varieties. Functionalized silica (e.g., amines, acids, thiols, etc.) can also be very effective for removing certain impurities such as Pd, Cu, Fe, etc.
Your goal should be to use all methods at your disposal to progressively clean up your mixture, step-by-step, and ultimately make crystallization easier to accomplish. For example, rather than “standard work up,” try washing with “standard work up” materials and analyze each layer and wash. If 5% brine is removing a significant amount of impurities, keep washing with 5% brine. Rather than just letting the organic layer stir for a few minutes with a drying agent before filtering, let the organic layer stir for 30 minutes with a drying agent and activated carbon (or functionalized silica) and then filter.
Figure 2. Common chiral and achiral salting reagents.
Goal
Your goal should be to create a scalable, reproducible, efficient, safe, and high yielding process so when there is a need for more of the material, it can be accomplished with relative efficiency and in a safe manner.
References
NRC (National Research Council). Prudent Practices in the Laboratory. Handling and Management of Chemical Hazards. National Academy Press: Washington, DC, 2011.
Furniss, B. S.; Hannaford, A. J.; Smith, P. W. G.; Tatchell, A. R. Vogel’s Textbook of Practical Organic Chemistry, 5th ed. Longman Group UK Limited: Singapore, 1989.
Anderson, N. G. Practical Process Research & Development, Elsevier: Waltham MA, 2000.
McConville, F. X. The Pilot Plant Real Book, FXM Engineering and Design: Worcester MA. 2006.
D. J. Am Ende (Ed). Chemical Engineering in the Pharmaceutical Industry: R & D to Manufacturing. John Wiley & Sons, Inc.: Hoboken, NJ 2011.
R. J. Alaimo (Ed). Handbook of Chemical Health and Safety. Oxford University Press: New York NY, 2001.